Amino Acids

Amino acids are the building blocks of proteins and play an important role in the human body for vital processes like cell formation and synthesis of hormones and neurotransmitters. There are 20 different amino acids required for the body to function properly, of which nine are essential. These essential amino acids are not synthesized in the body and must be ingested with our diet or by means of food supplements. Amino acids, same as all nutritional substances that are used in the manufacturing process of food and drinks need to be compliant with EU Regulation (EU) No 609/2013. Shimadzu is offering the complete solution for amino acid analysis including high sensitivity system configurations, comprehensive software and full application support.
HPLC is the most popular method for analyzing amino acids in food and beverages. As generally UV detection doesn’t offer sufficient sensitivity and selectivity for their detection, the most common procedure is derivatization of the analytes of interest to enable highly sensitive and selective analysis using fluorescence detection.
Derivatization can take place before or after separation, which both have some distinct advantages and disadvantages – the ideal method depends on the target analysis.
Pre-column Derivatization Method (pre-label method)
When using a pre-column derivatization method, amino acids are derivatized before injection, the labeled compounds are separated and detected. Figure 1 shows a scheme of pre-column amino acid derivatization reactions, using either O-phthalaldehyde (OPA) or 9-fluorenylmethyl chloroformate (FMOC), which both are well-known derivatization reagents that rapidly react with amino acids at room temperature.
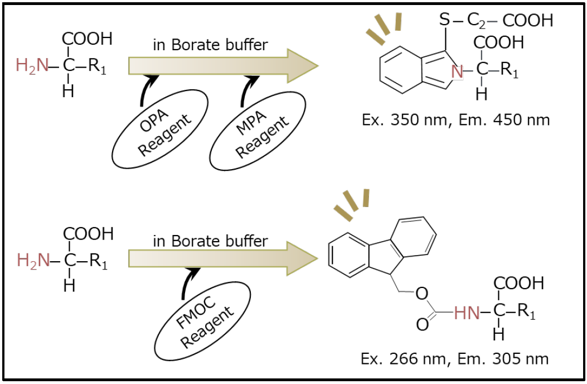
Figure 1: Scheme of Pre-column Amino Acid Derivatization Reactions
Top: Reaction with OPA Reagent | Bottom: Reaction with FMOC Reagent
The advantages of this method low consumption of reaction reagent, increased sensitivity when using quality reagents that provide very low background levels and even if unreacted derivatizing reagent is detected, as long as it is separated from the target compounds in the column, it does not cause a problem in the analysis.
On the other hand, a disadvantage is that because the derivatizing reagent is mixed directly with the sample, reaction efficiency (yield) is often influenced by sample matrix. Therefore, pre-column derivatization is the method of choice for analysis of a somewhat limited variety of samples with high sensitivity. Pre-column derivatization procedures vary widely, from simply mixing at room temperature, to reactions that require heating or protocols that require post-reaction cleanup. In many cases, reversed-phase chromatography is used to separate reaction products. Normally, reversed-phase chromatography is not well suited to separating highly-hydrophilic substances such as amino acids. However, as pre-column derivatization derivatizes samples before they are introduced to the column, amino acids can be modified with highly-hydrophobic functional groups to enable reversed-phase chromatography. Since reversed-phase methods provide excellent separation, it allows for fast analysis, as shown in an example in figure 2. It shows a chromatogram of analysis of 20 proteinogenic amino acids on an i-Series HPLC system using automated pre-column derivatization.
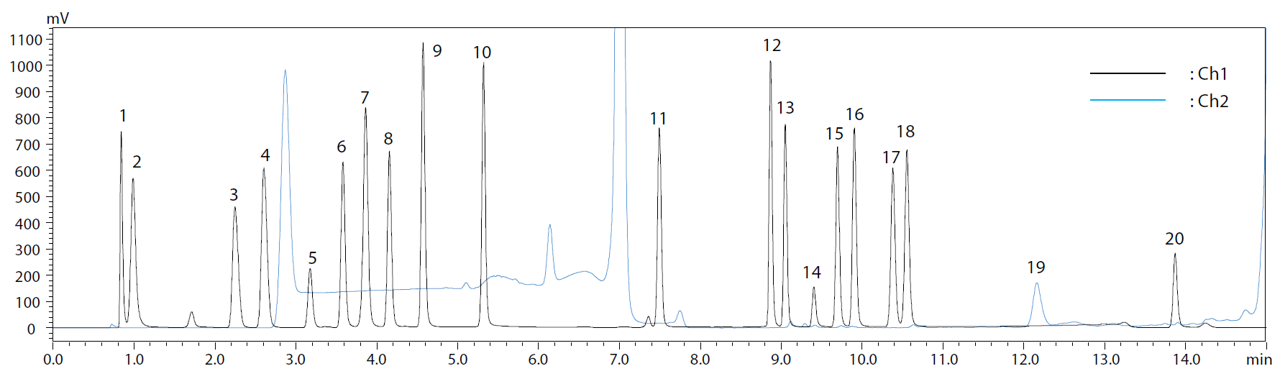
Figure 2: Chromatogram of analysis of 20 proteinogenic amino acids on an i-Series HPLC system using automated pre-column derivatization.
Post-column Derivatization Method (post-column reaction detection method)
The post-column derivatization method involves separating the amino acids in the column, then delivering and mixing the derivatizing reagent to let it react with the amino acids, before finally sending the products to the detector. A flow line diagram of a typical post-column derivatization process is shown in figure 3.
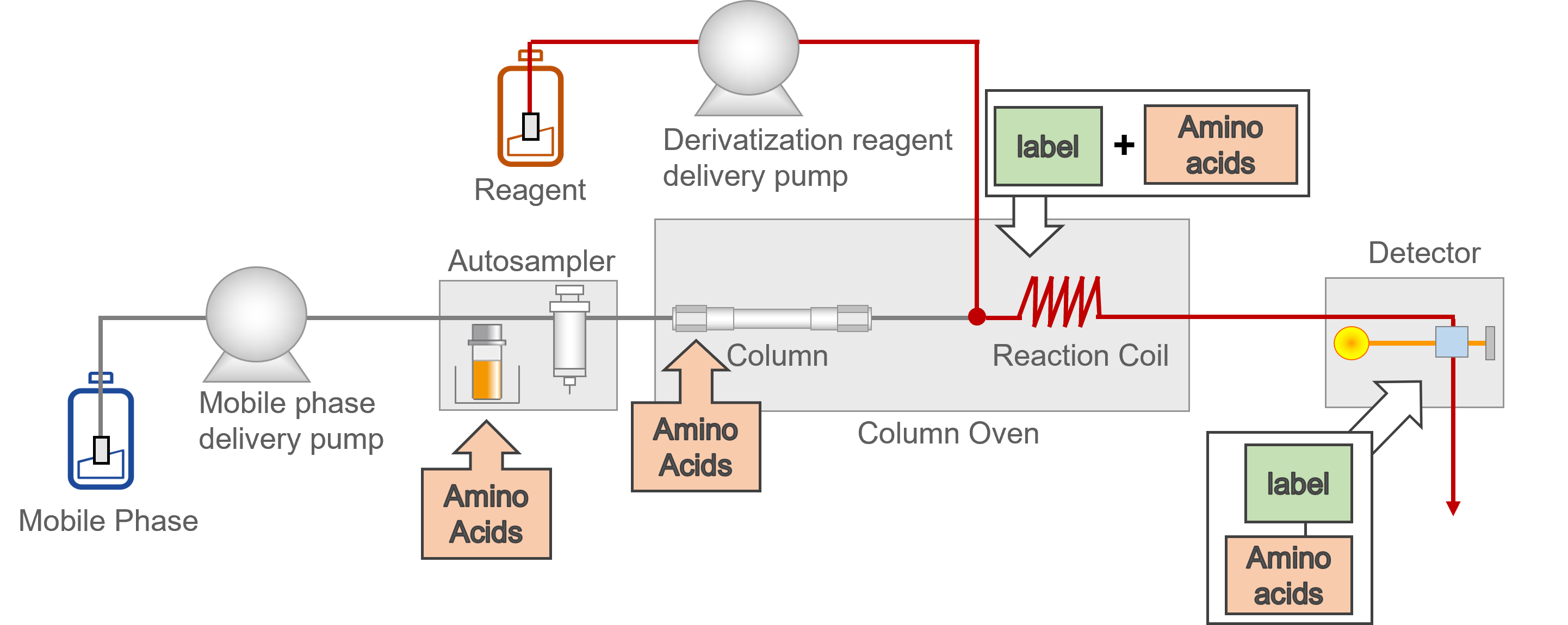
Figure 3: Flow line diagram the Nexera Amino Acid Analysis system using post-column derivatization
The advantages of this method are excellent quantitative performance and reproducibility. Since sample components are separated before the reaction, reaction efficiency is less prone to sample matrix effects, enabling its use for a wide range of samples in routine analysis. In contrast, its disadvantages include difficulty in increasing sensitivity and high consumption of reaction reagent, which is added in a constant flow, not permitting the detection of unreacted reagent.
The method most commonly used for separation is cation exchange chromatography. Amino acids are zwitterions that include both amino and carboxyl groups in its structure. Therefore, the higher the acidity of an amino acid the faster the elution, whereas the higher the basicity of an amino acid, the slower the elution. Therefore, cation exchange chromatography is able to easily and efficiently separate amino acids from each other and other typical amines. Figure 4 shows the chromatogram of the analysis of amino acids in protein powder using the Nexera Post-Column Amino Acid Analysis System.
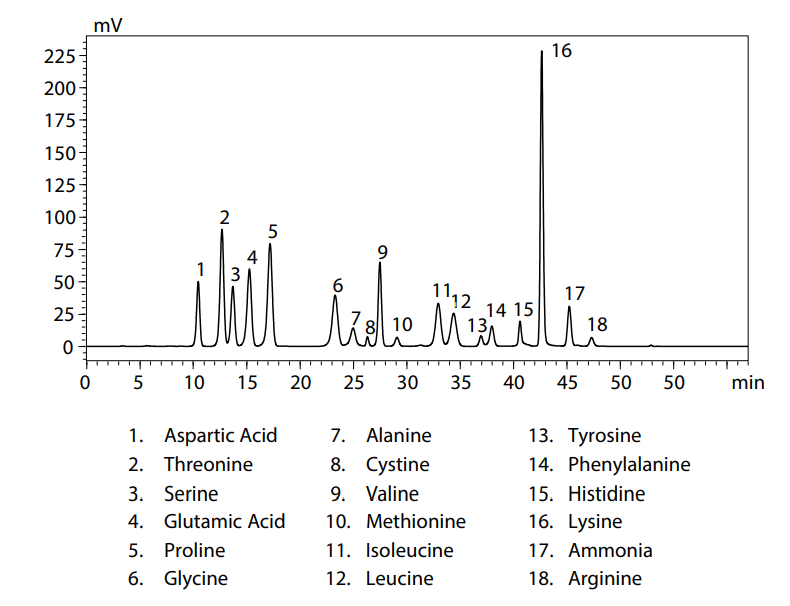
Figure 4: Chromatogram of the analysis of amino acids in protein powder using the Nexera Post-Column Amino Acid Analysis System
Comparison of Derivatization Methods for Amino Analysis
| Advantages | Disadvantages | |
|---|---|---|
| Pre-column Derivatization Method appropriate for higher-sensitivity analysis of a somewhat limited variety of samples, such as purified amino acids |
|
|
| Post-column Derivatization Once the reaction system is optimized, it can be used for a wide range of samples, including fermentation extracts, making it suitable for routine analysis with excellent quantitation performance |
|
|